Colaboración STERIS VHP y ChargePoint: Estudio de caso de Ritedose Corporation

Puntos clave:
- Ritedose utilizó la válvula ChargePoint AseptiSafe® Bio con el sistema STERIS VHP™ para la transferencia de polvo estéril en una sala limpia de grado C, evitando costosas actualizaciones de infraestructura.
- Se logró una reducción validada de 6 log mediante un ciclo de cuatro fases (deshumidificación, acondicionamiento, descontaminación y aireación), asegurando la esterilidad en el punto de transferencia.
- La solución integrada eliminó la necesidad de aisladores o RABS, lo que redujo los costos, el uso de energía y la complejidad.
- Los métodos de validación incluyeron indicadores químicos y biológicos, rellenos de medios y estudios de retención estéril.
- El sistema mejoró la eficiencia del proceso, el aseguramiento de esterilidad y la ergonomía, lo que demuestra beneficios escalables para la fabricación farmacéutica.
Ritedose Corporation es una organización norteamericana de desarrollo y fabricación por contrato (CDMO) que fabrica productos estériles en dosis unitarias.
Ritedose es una compañía farmacéutica de servicios integrales que aprovecha la tecnología de soplado-llenado-sellado. Sus capacidades se extienden mucho más allá de la fabricación, con un equipo de desarrollo interno que se especializa en todos los aspectos de llevar un producto al mercado, desde lotes a escala de laboratorio, presentaciones regulatorias, fabricación escalable y distribución. Cuentan con más de 20 años de experiencia en la fabricación de productos respiratorios y oftálmicos y unas instalaciones con una capacidad de 1700 millones de unidades que utilizan la última tecnología en formulación, soplado-llenado-sellado y envasado de alta velocidad.
Desafío
Ritedose Corporation quería resolver el problema de la carga de API estériles en un depósito de mezcla. Este es un problema común con todos los productos farmacéuticos preparados de manera aséptica.
Para el proceso era fundamental mantener las condiciones de esterilidad mientras se acoplaba un contenedor al recipiente y luego se transfería el API sólido para formar una suspensión líquida. Con un líquido totalmente disuelto, el producto podía filtrarse de manera estéril para garantizar su esterilidad al pasar al relleno. Aunque en este caso, el producto que se pasaba al relleno era una suspensión, por lo que esta opción no era posible.
Esto requería que todo el proceso se realizara bajo condiciones asépticas y, como tal, normalmente significaría que se requeriría una de las siguientes actualizaciones.
1. Actualizar toda la sala de una sala limpia de grado C a grado A.
2. Actualizar la sala a un entorno de grado B e introducir adicionalmente una zona sobrepresurizada de grado A alrededor del punto de llenado.
3. Actualizar la sala a un entorno de grado B e introducir adicionalmente un sistema RABS en el punto de llenado o recipiente lleno.
4. Mantener la sala limpia de grado C, pero introducir tecnología de aislamiento alrededor del punto de llenado o del recipiente lleno.
Tradicionalmente, los RAB y la tecnología de aisladores se habrían favorecido en este caso debido a las ventajas que aportan en términos de mejora del aseguramiento de esterilidad, empleando las técnicas fundamentales de separación y descontaminación. Dicho esto, al considerar algunas de las limitaciones negativas que conllevan estas tecnologías, como la elevada inversión de capital inicial, el espacio, la ergonomía y el costo y consumo de energía continuos, la empresa decidió buscar en otra parte una solución más adecuada para esta tarea crítica.
Solución
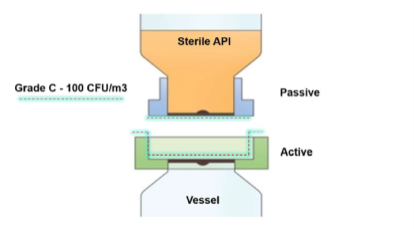
Se seleccionó un producto de válvula biológica aséptica como solución ideal a este problema, ya que proporciona una transferencia de polvo sellada en un pequeño espacio montado en el puerto de entrada del recipiente. La válvula se puede esterilizar previamente al vapor junto con el recipiente, a diferencia de las SBV tradicionales u otras conexiones convencionales (véase la ilustración 1a/b). En la conexión final, también eliminó de forma controlada y validada cualquier contaminación ambiental de las caras de contacto de la transferencia (véase la ilustración 2a/b).

Ilustración 1a de acoplamiento de SIP y tapa SIP

ilustración 1b de SIP a través de la tapa: activo preesterilizante y tapa
Transferencia de API estéril
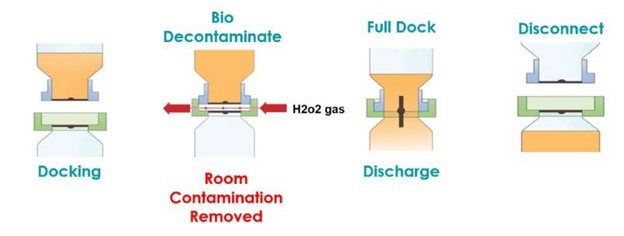
La válvula AseptiSafe Bio funciona creando una cámara sellada entre el recipiente de transferencia (sección pasiva) y el recipiente (sección activa). Cuando las dos mitades se acoplan, la cámara sellada se biodescontamina con un Sistema de biodescontaminación de peróxido de hidrógeno vaporizado, VHP®, de STERIS.
Ilustración 2a

La unidad STERIS VHP elimina cualquier contaminación biológica, hasta una reducción validada de 6 log, y deja el espacio y las caras de acoplamiento descontaminados y listos para acoplarse completamente. Una vez acoplado completamente, el disco puede abrirse, lo que permite transferir el producto del recipiente de transferencia al recipiente, sin riesgo de contaminación. Realizar esta transferencia aún dentro del espacio de grado C proporcionó enormes ventajas de costo y producción, aunque era necesario validar completamente el proceso para garantizar que las ventajas percibidas inicialmente pudieran demostrarse.
Ilustración 2b
Validación
El primer paso para validar microbiológicamente el proceso fue generar un ciclo de descontaminación validado para la fase de gasificación de peróxido de hidrógeno. El proceso seco y gaseoso de STERIS VHP consta de cuatro fases distintas, por las que pasará el generador para garantizar que se cumplen todas las condiciones críticas y que cada vez se realiza un ciclo validado. Las cuatro fases se establecen a tiempo.
1. Deshumidificación: donde la válvula que se gasifica reduce la humedad dentro de la cámara para proporcionar la condición ideal para la muerte biológica.
2. Acondicionamiento: donde se introduce VHP en la válvula para que se acumule hasta niveles que permitan lograr una buena descontaminación.
3. Descontaminación: la concentración de VHP se mantiene para desactivar cualquier actividad microbiológica dentro de la válvula.
4. Aireación: cuando se completa la descontaminación biológica, el VHP se retira del sistema para que no queden niveles nocivos de residuos. Normalmente el nivel de aceptación es de 1 ppm, aunque en este caso se utilizó 0.4 ppm como nivel de aceptación. Ritedose utilizó un límite de residuos más bajo para asegurarse de que disponían de un sistema sólido y de que no había posibilidad de contaminación del producto debido a residuos de gas.
El ciclo completo de descontaminación puede realizarse en tan solo cuatro minutos, aunque lo más habitual son 20 minutos. Para esta aplicación, el proceso solo se realizaba una vez al día y, para garantizar un ciclo sólido, se añadió tiempo adicional a cada una de las fases críticas, asegurando que se confirmaba la descontaminación y se aireaba el gas del sistema. Esto dio como resultado un ciclo completo de 41 minutos (acondicionamiento a aireación).
| Parámetro/fase | Deshumidificación | Acondicionamiento | Descontaminación | Aireación |
Tiempo, Mín | (10) | (0) | (6) | (25) |
Los ciclos iniciales utilizaron indicadores químicos (IQ) para determinar la distribución del H202. Cuando se lograron resultados satisfactorios de IQ, se introdujeron indicadores biológicos (IB) al proceso para confirmar que el proceso se logró con éxito. Al finalizar cada ciclo, se recogieron todos los IB e IQ. Luego se verificó el cambio de color en las tiras de IQ para asegurar una distribución uniforme del vapor. Los IB se transfirieron a un medio de cultivo adecuado, en este caso Medio de cultivo Spordex®, y se incubaron entre 55 °C y 60 °C durante siete días. Se observaron diariamente para cualquier crecimiento microbiano.

Los criterios de aceptación del ciclo incluían:
A) Todas las tiras de IQ utilizadas en el ciclo deben haber cambiado de color.
B) El IB de control positivo debe mostrar crecimiento.
C) Al menos un IB de cada ubicación no debe mostrar crecimiento.
Una vez desarrollado el ciclo, se ejecutó por triplicado para formar la calificación del rendimiento (CR) de este elemento del proceso.
Para validar completamente el sistema, el proceso se desafió con varias ejecuciones de medios antes de la validación. Estos exitosos desafíos de medios se llevaron a cabo en tres ejecuciones de medios en la CR. El mantenimiento de la esterilidad se demostró superior a 10 días con el producto transferido al recipiente y con la válvula biológica mantenida en la posición de enclavamiento cerrado. El periodo de retención estéril se demostró para la sección pasiva (producto en el recipiente de transferencia) durante 48 horas, lo que era más que adecuado, ya que normalmente sería como máximo la mitad de este tiempo.
Conclusión
La instalación ya está operativa y en plena producción. Las ventajas iniciales del proyecto, como el bajo costo de los equipos, el menor espacio ocupado y la facilidad de instalación, se han visto ahora compensadas por un mayor aseguramiento de esterilidad, la facilidad de uso para los usuarios y el bajo mantenimiento. El sistema es sencillo de usar, fácil de instalar/validar y ciertamente ha mejorado su proceso.
Uno de los aprendizajes de este proyecto se produjo en la fase de dispensación. En el momento de la validación, el sistema instalado era una solución reutilizable totalmente rígida en la que el API preesterilizado se suministraba a Ritedose en bolsas. Estas bolsas se abrieron y luego se subdividieron y dispensaron dentro de un aislador aséptico al recipiente de transferencia preesterilizado en autoclave y a la sección pasiva de la válvula biológica. Habría sido beneficioso esterilizar el producto, el recipiente y la conexión de transferencia en un solo paso (irradiación gamma), aunque debido a las limitaciones asociadas con la esterilización gamma de los ensamblajes de acero inoxidable y elastómeros como un solo artículo, esto no fue posible.
Esta solución ya está disponible en ChargePoint en forma de Pasivo de uso único (PUU)/ChargeBag® y en el futuro podría adoptarse para mejorar y agilizar el proceso. Por lo que todo el paquete (bolsa y pasivo) podría enviarse para la esterilización gamma, en lugar de tener varias etapas individuales de esterilización y ensamblaje aséptico.
«Era fundamental que eligiéramos la solución adecuada para este proyecto a fin de evitar la contaminación del producto y costosas pérdidas del mismo. Seleccionamos la válvula de transferencia ChargePoint AseptiSafe® Bio debido a su mayor aseguramiento de esterilidad al manipular ingredientes sensibles como nuestro principio activo. Hemos recibido un soporte considerable durante el proyecto y nos estamos beneficiando de importantes reducciones de costos y eficiencias de procesos».
Angie Koen, VP de Servicios Técnicos, The Ritedose Corporation.
Contenido sugerido

Guía para la implementación de un sistema de VHP para la biodescontaminación de instalaciones

Selección de la tecnología de descontaminación: En aerosol vs. VHP

Las ventajas de la descontaminación con peróxido de hidrógeno vaporizado
Únase a nuestra lista de correo electrónico para recibir las últimas novedades del sector y actualizaciones de productos.
Al hacer clic en Suscribirse, confirma que acepta nuestros Términos y condiciones.